Génomique
Génomique
La génomique est un domaine interdisciplinaire de la biologie axé sur la structure, la fonction, l'évolution, la cartographie et l'édition des génomes. Un génome est l'ensemble complet d'ADN d'un organisme, y compris tous ses gènes. Contrairement à la génétique, qui fait référence à l'étude des gènes individuels et de leurs rôles dans l'hérédité, la génomique vise à caractériser et quantifier collectivement tous les gènes d'un organisme, leurs interrelations et influence sur l'organisme. Les gènes peuvent diriger la production de protéines à l'aide d'enzymes et de molécules messagères. À leur tour, les protéines constituent les structures corporelles telles que les organes et les tissus, contrôlent les réactions chimiques et transportent des signaux entre les cellules. La génomique implique également le séquençage et l'analyse des génomes grâce à l'utilisation du séquençage d'ADN à haut débit et de la bioinformatique pour assembler et analyser la fonction et la structure de génomes entiers. Les progrès de la génomique ont déclenché une révolution dans la recherche basée sur la découverte et la biologie des systèmes pour faciliter la compréhension des systèmes biologiques les plus complexes tels que le cerveau.
Le domaine comprend également des études sur les phénomènes intragénomiques (dans le génome) comme l'épistasie (effet d'un gène sur un autre), la pléiotropie (un gène affectant plus d'un trait), l'hétérosis (vigueur hybride) et d'autres interactions entre les loci et les allèles dans le génome.
Table des matières
Histoire
Etymologie
Du grec ΓΕΝ gen , "gene" (gamma, epsilon, nu, epsilon) signifiant "devenir, create, creation, birth ", et les variantes suivantes: généalogie, genèse, génétique, génétique, génomère, génotype, genre etc. Alors que le mot génome (de l'allemand Genom , attribué à Hans Winkler) était en usage en anglais dès 1926, le terme génomique a été inventé par Tom Roderick, un généticien au Jackson Laboratory (Bar Harbor, Maine), au-dessus de la bière lors d'une réunion tenue je n Maryland sur la cartographie du génome humain en 1986.
Premiers efforts de séquençage
Suite à la confirmation par Rosalind Franklin de la structure hélicoïdale de l'ADN, James D. Watson et Francis Crick ont publié le structure de l'ADN en 1953 et la publication par Fred Sanger de la séquence d'acides aminés de l'insuline en 1955, le séquençage des acides nucléiques est devenu une cible majeure des premiers biologistes moléculaires. En 1964, Robert W. Holley et ses collègues ont publié la première séquence d'acide nucléique jamais déterminée, la séquence ribonucléotidique de l'ARN de transfert d'alanine. Prolongeant ce travail, Marshall Nirenberg et Philip Leder ont révélé la nature triplet du code génétique et ont pu déterminer les séquences de 54 codons sur 64 dans leurs expériences. En 1972, Walter Fiers et son équipe du Laboratoire de Biologie Moléculaire de l'Université de Gand (Gand, Belgique) ont été les premiers à déterminer la séquence d'un gène: le gène de la protéine d'enveloppe du bactériophage MS2. Le groupe de Fiers a développé son travail sur les protéines d'enveloppe MS2, déterminant la séquence nucléotidique complète du bactériophage MS2-ARN (dont le génome ne code que quatre gènes sur 3569 paires de bases) et du virus Simian 40 en 1976 et 1978, respectivement.
Développement de la technologie de séquençage de l'ADN
En plus de ses travaux fondamentaux sur la séquence d'acides aminés de l'insuline, Frederick Sanger et ses collègues ont joué un rôle clé dans le développement de techniques de séquençage de l'ADN qui ont permis la mise en place de projets de séquençage du génome. En 1975, lui et Alan Coulson ont publié une procédure de séquençage utilisant l'ADN polymérase avec des nucléotides radiomarqués qu'il a appelé la technique Plus et Minus . Cela impliquait deux méthodes étroitement liées qui ont généré des oligonucléotides courts avec des extrémités 3 'définies. Ceux-ci pourraient être fractionnés par électrophorèse sur un gel de polyacrylamide (appelé électrophorèse sur gel de polyacrylamide) et visualisés en utilisant une autoradiographie. La procédure pouvait séquencer jusqu'à 80 nucléotides en une seule fois et était une grande amélioration, mais était encore très laborieuse. Néanmoins, en 1977, son groupe a réussi à séquencer la plupart des 5 386 nucléotides du bactériophage simple brin φX174, complétant ainsi le premier génome à base d'ADN entièrement séquencé. Le raffinement de la méthode Plus et Moins a abouti à la méthode de terminaison de chaîne, ou méthode Sanger (voir ci-dessous), qui a formé la base des techniques de séquençage de l'ADN, de cartographie du génome, de stockage de données et d'analyse bioinformatique le plus largement utilisé dans le quart de siècle suivant de recherche. La même année, Walter Gilbert et Allan Maxam de l'Université de Harvard ont développé indépendamment la méthode Maxam-Gilbert (également connue sous le nom de méthode chimique ) de séquençage de l'ADN, impliquant le clivage préférentiel de l'ADN à des bases connues, un moins méthode efficace. Pour leur travail révolutionnaire dans le séquençage des acides nucléiques, Gilbert et Sanger ont partagé la moitié du prix Nobel de chimie 1980 avec Paul Berg (ADN recombinant).
Génomes complets
L'avènement de ces technologies a entraîné une intensification rapide de la portée et de la vitesse d'achèvement des projets de séquençage du génome. La première séquence génomique complète d'un organite eucaryote, la mitochondrie humaine (16 568 pb, environ 16,6 kb), a été signalée en 1981, et les premiers génomes chloroplastiques ont suivi en 1986. En 1992, le premier chromosome eucaryote, le chromosome III de la levure de bière Saccharomyces cerevisiae (315 kb) a été séquencé. Le premier organisme vivant libre à être séquencé a été celui de Haemophilus influenzae (1,8 Mo) en 1995. L'année suivante, un consortium de chercheurs de laboratoires d'Amérique du Nord, d'Europe et du Japon a annoncé l'achèvement du première séquence complète du génome d'un eucaryote, S. cerevisiae (12,1 Mb), et depuis lors, les génomes ont continué à être séquencés à un rythme de croissance exponentielle. Depuis octobre 2011, les séquences complètes sont disponibles pour: 2 719 virus, 1 115 archées et bactéries et 36 eucaryotes, dont environ la moitié sont des champignons.
La plupart des micro-organismes dont les génomes ont été complètement séquencés sont problématiques pathogènes, tels que Haemophilus influenzae , qui a entraîné un biais prononcé dans leur distribution phylogénétique par rapport à l’étendue de la diversité microbienne. Parmi les autres espèces séquencées, la plupart ont été choisies parce qu'elles étaient des organismes modèles bien étudiés ou promettaient de devenir de bons modèles. La levure ( Saccharomyces cerevisiae ) a longtemps été un organisme modèle important pour la cellule eucaryote, tandis que la mouche des fruits Drosophila melanogaster a été un outil très important (notamment au début de la phase pré-moléculaire la génétique). Le ver Caenorhabditis elegans est un modèle simple souvent utilisé pour les organismes multicellulaires. Le poisson zèbre Brachydanio rerio est utilisé pour de nombreuses études de développement au niveau moléculaire, et la plante Arabidopsis thaliana est un organisme modèle pour les plantes à fleurs. Le poisson-globe japonais ( Takifugu rubripes ) et le poisson-globe vert tacheté ( Tetraodon nigroviridis ) sont intéressants en raison de leurs génomes petits et compacts, qui contiennent très peu d'ADN non codant par rapport à la plupart des espèces . Le chien mammifère ( Canis familiaris ), le rat brun ( Rattus norvegicus ), la souris ( Mus musculus ) et le chimpanzé ( Pan troglodytes ) sont tous des animaux modèles importants dans la recherche médicale.
Une ébauche du génome humain a été achevée par le Human Genome Project au début de 2001, créant beaucoup de fanfare. Ce projet, achevé en 2003, a séquencé le génome entier pour une personne spécifique, et en 2007, cette séquence a été déclarée «terminée» (moins d'une erreur sur 20 000 bases et tous les chromosomes assemblés). Dans les années qui ont suivi, les génomes de nombreux autres individus ont été séquencés, en partie sous les auspices du 1000 Genomes Project, qui a annoncé le séquençage de 1 092 génomes en octobre 2012. L'achèvement de ce projet a été rendu possible par le développement de plus des technologies de séquençage efficaces et a nécessité l’engagement d’importantes ressources bioinformatiques issues d’une grande collaboration internationale. L'analyse continue des données génomiques humaines a de profondes répercussions politiques et sociales sur les sociétés humaines.
La révolution «omique»
Le néologisme anglophone omics fait référence de manière informelle à un domaine d'étude en la biologie se terminant par -omique , comme la génomique, la protéomique ou la métabolomique. Le suffixe associé -ome est utilisé pour aborder les objets d'étude de ces domaines, tels que le génome, le protéome ou le métabolome respectivement. Le suffixe -ome utilisé en biologie moléculaire se réfère à une totalité d'une certaine sorte; de même, l'omique en est venu à désigner généralement l'étude de grands ensembles de données biologiques complets. Si la croissance de l'utilisation du terme a conduit certains scientifiques (Jonathan Eisen, entre autres) à affirmer qu'il a été survendu, il reflète le changement d'orientation vers l'analyse quantitative de l'assortiment complet ou presque complet de tous les constituants de un système. Dans l'étude des symbioses, par exemple, les chercheurs qui étaient autrefois limités à l'étude d'un seul produit génique peuvent désormais comparer simultanément le complément total de plusieurs types de molécules biologiques.
Analyse du génome
Une fois qu'un organisme a été sélectionné, les projets génomiques impliquent trois éléments: le séquençage de l'ADN, l'assemblage de cette séquence pour créer une représentation du chromosome d'origine, et l'annotation et l'analyse de cette représentation.
Séquençage
Historiquement, le séquençage était effectué dans des centres de séquençage , des installations centralisées (allant de grandes institutions indépendantes telles que le Joint Genome Institute qui séquencent des dizaines de térabases par an, aux centres de recherche locaux de biologie moléculaire) laboratoires dotés de l’instrumentation coûteuse et du soutien technique nécessaire. Cependant, à mesure que la technologie de séquençage continue de s'améliorer, une nouvelle génération de séquenceurs de paillasse efficaces à rotation rapide est à la portée du laboratoire universitaire moyen. Dans l'ensemble, les approches de séquençage du génome se divisent en deux grandes catégories, le séquençage fusil de chasse et haut débit (ou nouvelle génération ).
Le séquençage Shotgun est une méthode de séquençage conçue pour l'analyse de séquences d'ADN de plus de 1000 paires de bases, jusqu'à et y compris des chromosomes entiers. Il est nommé par analogie avec le schéma de tir quasi aléatoire et en expansion rapide d'un fusil de chasse. Étant donné que le séquençage par électrophorèse sur gel ne peut être utilisé que pour des séquences assez courtes (100 à 1000 paires de bases), des séquences d'ADN plus longues doivent être divisées en petits segments aléatoires qui sont ensuite séquencés pour obtenir des lectures . Plusieurs lectures se chevauchant pour l'ADN cible sont obtenues en effectuant plusieurs tours de cette fragmentation et séquençage. Les programmes informatiques utilisent ensuite les extrémités qui se chevauchent de différentes lectures pour les assembler en une séquence continue. Le séquençage par fusil de chasse est un processus d'échantillonnage aléatoire, nécessitant un suréchantillonnage pour s'assurer qu'un nucléotide donné est représenté dans la séquence reconstruite; le nombre moyen de lectures par lesquelles un génome est suréchantillonné est appelé couverture.
Pendant une grande partie de son histoire, la technologie sous-jacente au séquençage des fusils de chasse était la méthode classique de terminaison de chaîne ou «méthode Sanger», qui est basé sur l'incorporation sélective de didésoxynucléotides de terminaison de chaîne par l'ADN polymérase pendant la réplication d'ADN in vitro. Récemment, le séquençage des fusils de chasse a été supplanté par des méthodes de séquençage à haut débit, en particulier pour les analyses génomiques automatisées à grande échelle. Cependant, la méthode Sanger reste largement utilisée, principalement pour des projets à plus petite échelle et pour obtenir des lectures de séquences d'ADN contiguës particulièrement longues (& gt; 500 nucléotides). Les méthodes de terminaison de chaîne nécessitent une matrice d'ADN simple brin, une amorce d'ADN, une ADN polymérase, des désoxynucléosidétriphosphates normaux (dNTP) et des nucléotides modifiés (didésoxyNTP) qui terminent l'élongation du brin d'ADN. Ces nucléotides de terminaison de chaîne sont dépourvus d'un groupe 3'-OH requis pour la formation d'une liaison phosphodiester entre deux nucléotides, amenant l'ADN polymérase à cesser l'extension de l'ADN lorsqu'un ddNTP est incorporé. Les ddNTP peuvent être marqués radioactivement ou par fluorescence pour une détection dans des séquenceurs d'ADN. En règle générale, ces machines peuvent séquencer jusqu'à 96 échantillons d'ADN en un seul lot (analyse) en jusqu'à 48 cycles par jour.
La forte demande de séquençage à faible coût a conduit au développement du séquençage à haut débit technologies qui parallélisent le processus de séquençage, produisant des milliers ou des millions de séquences à la fois. Le séquençage à haut débit vise à réduire le coût du séquençage de l'ADN au-delà de ce qui est possible avec les méthodes standard de terminateur de colorant. Dans le séquençage à très haut débit, jusqu'à 500 000 opérations de séquençage par synthèse peuvent être exécutées en parallèle.
La méthode de séquençage de colorant Illumina est basée sur des terminateurs de colorant réversibles et a été développée en 1996 au Institut de recherche biomédicale de Genève, par Pascal Mayer et Laurent Farinelli. Dans ce procédé, les molécules d'ADN et les amorces sont d'abord fixées sur une lame et amplifiées avec de la polymérase de sorte que des colonies clonales locales, initialement appelées "colonies d'ADN", sont formées. Pour déterminer la séquence, quatre types de bases de terminaison réversibles (bases RT) sont ajoutés et les nucléotides non incorporés sont éliminés par lavage. Contrairement au pyroséquençage, les chaînes d'ADN sont étendues un nucléotide à la fois et l'acquisition d'image peut être effectuée à un moment retardé, ce qui permet de capturer de très grands tableaux de colonies d'ADN par des images séquentielles prises à partir d'une seule caméra. Le découplage de la réaction enzymatique et de la capture d'image permet un débit optimal et une capacité de séquençage théoriquement illimitée; avec une configuration optimale, le débit ultime de l'instrument dépend uniquement du taux de conversion A / N de la caméra. La caméra prend des images des nucléotides marqués par fluorescence, puis le colorant avec le bloqueur terminal 3 'est chimiquement éliminé de l'ADN, permettant le cycle suivant.
Une approche alternative, le séquençage des semi-conducteurs ioniques, est basée sur la chimie standard de réplication de l'ADN. Cette technologie mesure la libération d'un ion hydrogène à chaque fois qu'une base est incorporée. Un micropuits contenant l'ADN matrice est inondé d'un seul nucléotide, si le nucléotide est complémentaire du brin matrice, il sera incorporé et un ion hydrogène sera libéré. Cette version déclenche un capteur d'ions ISFET. Si un homopolymère est présent dans la séquence modèle, plusieurs nucléotides seront incorporés dans un seul cycle d'inondation, et le signal électrique détecté sera proportionnellement plus élevé.
Assemblage
L'assemblage de séquence fait référence à l'alignement et fusionner des fragments d'une séquence d'ADN beaucoup plus longue afin de reconstruire la séquence originale. Cela est nécessaire car la technologie actuelle de séquençage de l'ADN ne peut pas lire des génomes entiers comme une séquence continue, mais lit plutôt de petits morceaux de 20 à 1000 bases, selon la technologie utilisée. Les technologies de séquençage de troisième génération telles que PacBio ou Oxford Nanopore génèrent régulièrement des lectures de séquençage> 10 ko de longueur; cependant, ils ont un taux d'erreur élevé d'environ 15 pour cent. En général, les fragments courts, appelés lectures, résultent du séquençage de l'ADN génomique par fusil de chasse ou des transcriptions de gènes (EST).
L'assemblage peut être globalement catégorisé en deux approches: l'assemblage de novo , pour génomes qui ne sont similaires à aucun autre séquencé dans le passé, et assemblage comparatif, qui utilise la séquence existante d'un organisme étroitement apparenté comme référence lors de l'assemblage. Par rapport à l'assemblage comparatif, l'assemblage de novo est difficile en termes de calcul (NP-hard), ce qui le rend moins favorable pour les technologies NGS à lecture courte. Dans le paradigme d'assemblage de novo , il existe deux stratégies principales pour l'assemblage, les stratégies de chemin eulérien et les stratégies de chevauchement-disposition-consensus (OLC). Les stratégies OLC essaient finalement de créer un chemin hamiltonien à travers un graphe de chevauchement qui est un problème NP-difficile. Les stratégies de chemin eulérien sont plus faciles à traiter, car elles essaient de trouver un chemin eulérien à travers un graphe deBruijn.
Les génomes finis sont définis comme ayant une seule séquence contiguë sans ambiguïtés représentant chaque réplicon.
Annotation
L'assemblage de séquences d'ADN seul est de peu de valeur sans analyse supplémentaire. L'annotation du génome est le processus qui consiste à attacher des informations biologiques à des séquences et se compose de trois étapes principales:
- identifier les parties du génome qui ne codent pas pour les protéines
- identifier les éléments sur le génome, un processus appelé prédiction génétique, et
- attacher des informations biologiques à ces éléments.
Les outils d'annotation automatique tentent d'effectuer ces étapes in silico , par opposition à l'annotation manuelle (aka curation) qui implique l'expertise humaine et la vérification expérimentale potentielle. Idéalement, ces approches coexistent et se complètent dans le même pipeline d'annotations (voir également ci-dessous).
Traditionnellement, le niveau de base de l'annotation consiste à utiliser BLAST pour trouver des similitudes, puis à annoter des génomes basés sur des homologues . Plus récemment, des informations supplémentaires ont été ajoutées à la plate-forme d'annotation. Les informations supplémentaires permettent aux annotateurs manuels de déconvoluer les divergences entre les gènes qui reçoivent la même annotation. Certaines bases de données utilisent des informations de contexte génomique, des scores de similarité, des données expérimentales et des intégrations d'autres ressources pour fournir des annotations génomiques via leur approche des sous-systèmes. D'autres bases de données (par exemple Ensembl) s'appuient à la fois sur des sources de données organisées et sur une gamme d'outils logiciels dans leur pipeline d'annotation automatisée du génome. L'annotation structurelle consiste en l'identification des éléments génomiques, principalement les ORF et leur localisation, ou structure du gène. L'annotation fonctionnelle consiste à attacher des informations biologiques à des éléments génomiques.
Séquençage des pipelines et des bases de données
Le besoin de reproductibilité et de gestion efficace de la grande quantité de données associées avec des projets génomiques signifient que les pipelines de calcul ont des applications importantes en génomique.
Domaines de recherche
Génomique fonctionnelle
La génomique fonctionnelle est un domaine de la biologie moléculaire qui tente d'utiliser la vaste richesse de données produites par des projets génomiques (tels que des projets de séquençage du génome) pour décrire les fonctions et les interactions des gènes (et des protéines). La génomique fonctionnelle se concentre sur les aspects dynamiques tels que la transcription génique, la traduction et les interactions protéine-protéine, par opposition aux aspects statiques de l'information génomique comme la séquence ou les structures d'ADN. La génomique fonctionnelle tente de répondre aux questions sur la fonction de l'ADN au niveau des gènes, des transcrits d'ARN et des produits protéiques. Une caractéristique clé des études de génomique fonctionnelle est leur approche à l'échelle du génome de ces questions, impliquant généralement des méthodes à haut débit plutôt qu'une approche plus traditionnelle «gène par gène».
Une branche majeure de la génomique est toujours préoccupé par le séquençage des génomes de divers organismes, mais la connaissance des génomes complets a créé la possibilité pour le domaine de la génomique fonctionnelle, principalement concerné par les modèles d'expression des gènes dans diverses conditions. Les outils les plus importants ici sont les puces à ADN et la bioinformatique.
Génomique structurale
La génomique structurale cherche à décrire la structure tridimensionnelle de chaque protéine codée par un génome donné. Cette approche basée sur le génome permet une méthode à haut débit de détermination de la structure par une combinaison d'approches expérimentales et de modélisation. La principale différence entre la génomique structurale et la prédiction structurale traditionnelle est que la génomique structurelle tente de déterminer la structure de chaque protéine codée par le génome, plutôt que de se concentrer sur une protéine particulière. Avec des séquences génomiques complètes disponibles, la prédiction de structure peut être effectuée plus rapidement grâce à une combinaison d'approches expérimentales et de modélisation, en particulier parce que la disponibilité d'un grand nombre de génomes séquencés et de structures protéiques précédemment résolues permet aux scientifiques de modéliser la structure des protéines sur les structures déjà résolues. homologues. La génomique structurelle implique l'adoption d'un grand nombre d'approches pour la détermination de la structure, y compris des méthodes expérimentales utilisant des séquences génomiques ou des approches basées sur la modélisation basées sur une séquence ou une homologie structurale à une protéine de structure connue ou basées sur des principes chimiques et physiques pour une protéine sans homologie à toute structure connue. Contrairement à la biologie structurale traditionnelle, la détermination d'une structure protéique à travers un effort de génomique structurale intervient souvent (mais pas toujours) avant que l'on ne sache quoi que ce soit concernant la fonction protéique. Cela pose de nouveaux défis en bioinformatique structurale, c'est-à-dire en déterminant la fonction d'une protéine à partir de sa structure 3D.
L'épigénomique
L'épigénomique est l'étude de l'ensemble complet des modifications épigénétiques sur le matériel génétique d'une cellule , connu sous le nom d'épigénome. Les modifications épigénétiques sont des modifications réversibles de l'ADN ou des histones d'une cellule qui affectent l'expression génique sans altérer la séquence d'ADN (Russell 2010 p. 475). Deux des modifications épigénétiques les plus caractérisées sont la méthylation de l'ADN et la modification des histones. Les modifications épigénétiques jouent un rôle important dans l'expression et la régulation des gènes, et sont impliquées dans de nombreux processus cellulaires tels que la différenciation / développement et la tumorigenèse. L'étude de l'épigénétique au niveau mondial n'a été rendue possible que récemment grâce à l'adaptation de tests génomiques à haut débit.
La métagénomique
La métagénomique est l'étude des métagénomes , matériel génétique récupéré directement à partir d'échantillons environnementaux. Le vaste domaine peut également être appelé génomique environnementale, écogénomique ou génomique communautaire. Alors que la microbiologie traditionnelle et le séquençage du génome microbien reposent sur des cultures clonales cultivées, le séquençage précoce des gènes environnementaux a cloné des gènes spécifiques (souvent le gène de l'ARNr 16S) pour produire un profil de diversité dans un échantillon naturel. Ces travaux ont révélé que la grande majorité de la biodiversité microbienne avait été manquée par les méthodes basées sur la culture. Des études récentes utilisent le séquençage Sanger «fusil à pompe» ou le pyroséquençage massivement parallèle pour obtenir des échantillons largement non biaisés de tous les gènes de tous les membres des communautés échantillonnées. En raison de son pouvoir de révéler la diversité jusque-là cachée de la vie microscopique, la métagénomique offre un objectif puissant pour visualiser le monde microbien qui a le potentiel de révolutionner la compréhension du monde vivant dans son ensemble.
Systèmes modèles
Les bactériophages ont joué et continuent de jouer un rôle clé dans la génétique bactérienne et la biologie moléculaire. Historiquement, ils ont été utilisés pour définir la structure des gènes et la régulation des gènes. Le premier génome séquencé était également un bactériophage. Cependant, la recherche sur les bactériophages n'a pas conduit la révolution de la génomique, qui est clairement dominée par la génomique bactérienne. Ce n'est que très récemment que l'étude des génomes des bactériophages est devenue importante, permettant ainsi aux chercheurs de comprendre les mécanismes sous-jacents à l'évolution des phages. Les séquences du génome des bactériophages peuvent être obtenues par séquençage direct de bactériophages isolés, mais peuvent également être dérivées dans le cadre de génomes microbiens. L'analyse des génomes bactériens a montré qu'une quantité substantielle d'ADN microbien se compose de séquences de prophage et d'éléments de type prophage. L'exploration détaillée de ces séquences dans une base de données permet de mieux comprendre le rôle des prophages dans la formation du génome bactérien: dans l'ensemble, cette méthode a vérifié de nombreux groupes bactériophages connus, ce qui en fait un outil utile pour prédire les relations des prophages à partir de génomes bactériens.
Il existe actuellement 24 cyanobactéries pour lesquelles une séquence génomique totale est disponible. 15 de ces cyanobactéries proviennent du milieu marin. Il s'agit de six souches de Prochlorococcus , de sept souches de Synechococcus marines, de Trichodesmium erythraeum IMS101 et de Crocosphaera watsonii WH8501. Plusieurs études ont montré comment ces séquences pouvaient être utilisées avec beaucoup de succès pour déduire d'importantes caractéristiques écologiques et physiologiques des cyanobactéries marines. Cependant, de nombreux autres projets génomiques sont actuellement en cours, parmi lesquels d’autres isolats de Prochlorococcus et de Synechococcus marins, Acaryochloris et Prochloron , les cyanobactéries filamenteuses fixatrices de N2 Nodularia spumigena , Lyngbya aestuarii et Lyngbya majuscula , ainsi que les bactériophages infectant la cyanobacère marine. Ainsi, le volume croissant d'informations sur le génome peut également être exploité d'une manière plus générale pour résoudre des problèmes mondiaux en appliquant une approche comparative. Quelques exemples nouveaux et passionnants de progrès dans ce domaine sont l'identification de gènes pour les ARN régulateurs, des aperçus sur l'origine évolutive de la photosynthèse ou l'estimation de la contribution du transfert horizontal de gènes aux génomes qui ont été analysés.
Applications de la génomique
La génomique a fourni des applications dans de nombreux domaines, notamment la médecine, la biotechnologie, l'anthropologie et d'autres sciences sociales.
Médecine génomique
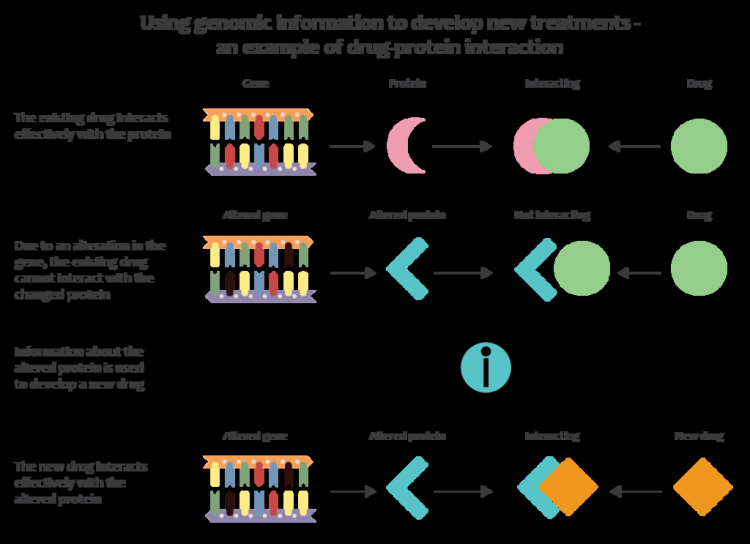
Génomique de nouvelle génération Les technologies permettent aux cliniciens et aux chercheurs biomédicaux d'augmenter considérablement la quantité de données génomiques collectées sur de grandes populations étudiées. Lorsqu'il est combiné avec de nouvelles approches informatiques qui intègrent de nombreux types de données avec des données génomiques dans la recherche sur les maladies, cela permet aux chercheurs de mieux comprendre les bases génétiques de la réponse aux médicaments et de la maladie. Les premiers efforts pour appliquer le génome à la médecine ont inclus ceux d'une équipe de Stanford dirigée par Euan Ashley qui a développé les premiers outils pour l'interprétation médicale d'un génome humain. Par exemple, le programme de recherche All of Us vise à collecter des données de séquence génomique auprès d'un million de participants pour devenir un élément essentiel de la plate-forme de recherche en médecine de précision.
Biologie synthétique et bio-ingénierie
La croissance des connaissances génomiques a permis des applications de plus en plus sophistiquées de la biologie synthétique. En 2010, des chercheurs de l'Institut J. Craig Venter ont annoncé la création d'une espèce partiellement synthétique de bactérie, Mycoplasma laboratorium , dérivée du génome de Mycoplasma genitalium .
Génomique de la conservation
Les écologistes peuvent utiliser les informations recueillies par séquençage génomique afin de mieux évaluer les facteurs génétiques clés de la conservation des espèces, tels que la diversité génétique d'une population ou si un individu est hétérozygote pour un récessif trouble génétique héréditaire. En utilisant des données génomiques pour évaluer les effets des processus évolutifs et pour détecter des modèles de variation dans une population donnée, les défenseurs de l'environnement peuvent formuler des plans pour aider une espèce donnée sans autant de variables inconnues que celles qui ne sont pas traitées par les approches génétiques standard.

Gaten Matarazzo subit une quatrième intervention chirurgicale pour un trouble osseux rare, la dysplasie cléidocrânienne - mais qu'est-ce que c'est?
La star de Stranger Things, Gaten Matarazzo, a révélé mercredi qu'il avait subi …

George W. Citroner
George W. Citroner couvre les dernières nouvelles en médecine et en santé. Il …
